プロテインキナーゼは、ほぼすべての細胞活動の調節において重要な役割を担います。これらのシグナル伝達酵素は、リン酸化を介して、タンパク質 (および他のキナーゼ) の活性や安定性を制御し、細胞内での局在を変化させながら、または破壊対象をマークするなどし、細胞の増殖や分裂、代謝をまるでオーケストラの指揮者のように見事に調節します。
そのため、これらの重要な調節タンパク質の調節不全が疾患、特にがんや神経疾患、発達障害などに共通する機構であることは何ら不思議ではありません。しかしながら、ヒトゲノムには530種以上のキナーゼが存在し、そのうちの400種以上が触媒活性を持つことが分かっており、どのキナーゼが標的タンパク質 (または対応するキナーゼ基質) を調節するかを特定することは困難、というよりむしろ不可能です。
重要な基質の調節不全を引き起こすキナーゼを簡単に特定することができれば、多くの新たな薬物標的を発見し、治療環境を一変することができると以前から分かっていましたが、現在に至るまで、この単純な作業を大規模に実施することは不可能でした。
しかし、MITやイェール大学医学部、ワイル・コーネル医科大学の研究者が、強力な計算ツールや精製されたキナーゼとペプチドライブラリー、そしてCell Signaling Technologyが提供するリソースであるPhosphositePlusから得られるデータを活用し、特定のタンパク質基質の制御を担うキナーゼを予測するアルゴリズムを開発しました。
The Kinase Libraryという名のそのツールは、現在、PhoshoSitePlusよりβ版が無料で公開されています。
がんにおけるセリン/スレオニンキナーゼの活性化状態に焦点を当てたこのプロジェクトの詳細を記したAn atlas of substrate specificities for human serine/threonine kinomeと題する論文は、2023年1月にNature誌に投稿されました1。本ブログ記事では、このプロジェクトに貢献した画期的な研究と、CSTとの共同により作成されたThe Kinase Libraryが、研究者による新たな薬物標的の発見にどのように役立つかを紹介します。
生涯にわたる探求:シグナル伝達経路におけるプロテインキナーゼの解明
ハーバード大学医学部とダナファーバー研究所に所属するLewis (Lew) Cantley教授は、ワイル・コーネル医科大学の教授であった際に本研究を行っており、何十年も前からキナーゼのシグナル伝達に関心をもっていました。Cantley教授による、PI3キナーゼなどのシグナル伝達と代謝における画期的な発見は有名であり、がんの生物学と疾患進行の理解に大きな影響を与えました。
何十年にもわたり、Cantley教授らのプロテオミクスの研究者は、健康な細胞と比較して、がん細胞でより高いリン酸化レベルがみられるタンパク質の同定に質量分析 (MS) を用いていました。しかし、MSをベースとするリン酸化プロテオミクスからは、リン酸化されたタンパク質の情報しか得られず、これらのリン酸化イベントを引き起こす直接的なキナーゼを特定することはできません。さらに、潜在的なリン酸化部位の特定にMSデータを用いる際に、しばしばそのデータには数百から数万に及ぶあらゆる翻訳後修飾 (PTM) サイトやモチーフが含まれている場合があり、個々のキナーゼの特異性の確認が難しくなります。これまでに、ヒトのタンパク質では数万ものセリンとスレオニンのリン酸化部位が特定されており、そのうちの数千がヒトの疾患や生物学的プロセスに関連していることが分かっています。しかし、これらの修飾を担うキナーゼは、セリン/スレオニンモチーフの4%未満しか特定されていません。
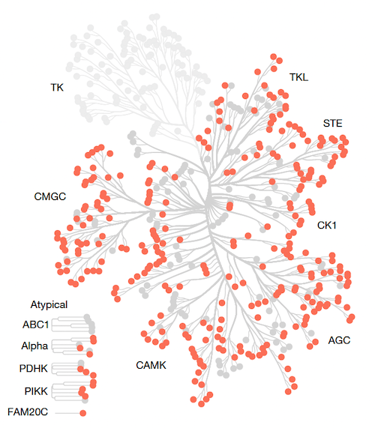
研究で解明されたセリン/スレオニンキナーゼに着目した、ヒトタンパク質キノームの樹状図1
しかし、現代の計算ツールは、従来の技術では得られなかった知見を提供してくれます。Cantley教授は、正しいデータを得ることができれば、タンパク質上のリン酸化部位の構造を活用し、特定のタンパク質に相互作用する既知のキナーゼの予測アルゴリズムの開発が可能であることを理解していました。ただし、そのようなアルゴリズムを作成するためにはまず、主要な既知のヒトキナーゼの結合の優先順を決定し、予測モデルを構築する必要がありました。
結合の優先順を決定するためのセリン/スレオニンキナーゼの精製
25年以上前、Nature誌に投稿した論文の他の責任著者であるMIT精密がん医療センターのdirectorを務めるMichael Yaffe氏とイェール大学医学部で薬学の准教授を務めるBenjamin Turk氏が、Cantley教授の研究室でこの偉大なプロジェクトを開始しました。この3人の研究者は数十年にわたり、キナーゼを研究し、そのシグナル伝達経路を解明するための新しい方法を開発してきました。
この3人の研究者による最初の飛躍的な発明のうちの1つが、様々なキナーゼと相互作用するモチーフを特定するためのペプチドライブラリーの開発でした。このモチーフライブラリーは、ペプチドモチーフとして機能する可能性があるすべてのアミノ酸配列を含む、約25億種類のペプチド基質で構成されています。リン酸化を例としてより詳しく説明します。このペプチドライブラリーは、アミノ酸配列内の単一のリン酸化ペプチドを固定し、その周囲を様々なアミノ酸に変化さて作製されています。その結果、特定のキナーゼが相互作用する可能性があるすべてのモチーフを含んだライブラリーが完成します。次に、ペプチドライブラリーに対し、目的のキナーゼと放射性標識したATP (ガンマP32) を用いたin vitroのリン酸化アッセイを行います。これらのペプチドアレイから得られるリン酸化の量から、キナーゼの標的となるセリンやスレオニン、またはチロシン部位の周囲において、キナーゼが優先する、または優先しない20個のアミノ酸に関する正確な情報が得られます。
Nature誌に投稿した論文の筆頭筆者であるワイル・コーネル医科大学の薬理学講師を務めるJared Johnson氏と、同大学の元大学院生であるTomer Yaron氏が、この研究で対象としているキナーゼの、基質への結合の優先順の決定を行いました。Cantley教授のペプチドライブラリーを用いて、ヒトで活性があると予測されるキナーゼの84%を占める、303種の既知のセリン/スレオニンキナーゼの結合の優先順の決定を率先して行いました。
この実験では、個々のキナーゼに対する組換え処理や、ペプチドアレイを用いた活性の測定も行なっています。この方法により、リン酸化部位の周辺のアミノ酸配列をベースとして、キナーゼがより強くリン酸化を優先する基質を得ることができました。
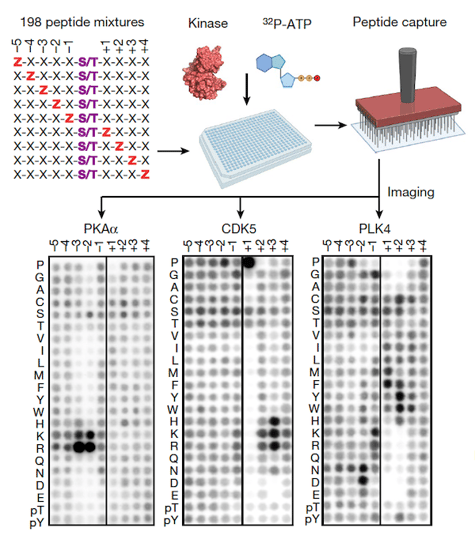 ヒトのセリン/スレオニンキノームの基質特異性の解析。PSPA解析の実験ワークフローと代表的な結果を示しています。Zは、20種類の天然アミノ酸またはリン酸化されたスレオニン (pThr) またはチロシン (pTyr) のいずれかの固定位置を示しています。Xは、固定されていないセリン、スレオニン、システイン以外のランダムなすべての天然アミノ酸の位置を示しています。●印は、優先される残基を示しています。図はBioRenderで作成しました1。
ヒトのセリン/スレオニンキノームの基質特異性の解析。PSPA解析の実験ワークフローと代表的な結果を示しています。Zは、20種類の天然アミノ酸またはリン酸化されたスレオニン (pThr) またはチロシン (pTyr) のいずれかの固定位置を示しています。Xは、固定されていないセリン、スレオニン、システイン以外のランダムなすべての天然アミノ酸の位置を示しています。●印は、優先される残基を示しています。図はBioRenderで作成しました1。
既知のセリン/スレオニンキナーゼに対するデータを利用し、キナーゼが優先して結合する基質を予測することができる計算モデルが開発されました。研究者は、各キナーゼ - ペプチドペアをスコアリングするために、正規化および確率を用いてスケーリングした大規模なデータセットを活用し、基質としての可能性がある、あらゆるアミノ酸配列に適用可能なスコアリング指標を開発しました。彼らは、キナーゼと基質ペプチドとの間のスコアを、数万もの部位を含むバックグラウンドのリン酸化プロテオームで割ることにより、各キナーゼ-ペプチドペアの優先スコアを百分率で算出しました。優先スコアは、キナーゼが、細胞内の他のすべての潜在的な基質と比較して、特定の基質をどの程度優先するのかを示します。
「リン酸化プロテオミクスの再興」
驚くことに、研究者は非常に異なるアミノ酸配列を持つタンパク質キナーゼが、類似した基質モチーフを持つタンパク質をリン酸化する場合が多々あることを発見しました。さらに、研究チームは、研究対象となるセリン/スレオニンキナーゼの約半数が、3つの主要なモチーフのいずれかを標的とすることを発見しました。残りの半数は、約12種のより小さなクラスのモチーフに特異的です。
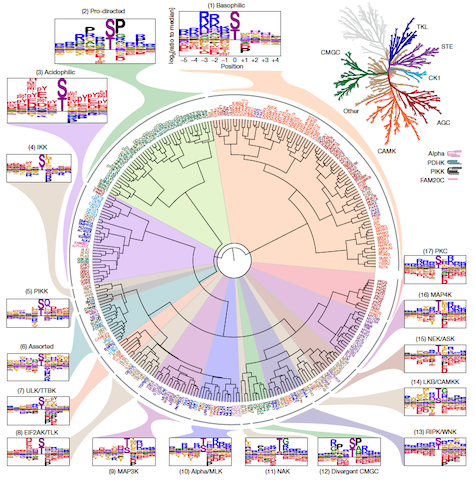
ヒトのセリン/スレオニンキノームにおけるリン酸化部位のモチーフツリー。303種のセリン/スレオニンキナーゼをアミノ酸モチーフ に基づき階層的クラスタリングを行いました (PSSM)。キナーゼ名は系統に応じて色分けしています (右上)2。フルサイズ画像はこちらの科学論文をご覧ください1。
研究チームは、本研究では標的化していなかった公開済みのリン酸化プロテオミクスのデータセットを用いて、予測システムを試験しました。1つの例として、彼らはPlk1と呼ばれ、細胞の増殖を調節するキナーゼに対する、抗がん作用をもつ阻害剤で処理した細胞のデータを解析しました。データセットから同定されたリン酸化部位のアミノ酸配列のみをベースとし、背後にある生物学的な情報を用いることなく試験したところ、リン酸化イベントの抑制状態に最も関連すると予測されるキナーゼとして、Plk1を特定することができました。さらに、さらなるキナーゼ群が関与する、より離れた下流の経路の解明に成功し、この予測システムが、複雑な世界中のリン酸化プロテオミクスのデータをまとめ上げることができることが明らかになりました。
この計算モデルは現在、The Kinase LibraryのPhosphositeに採用されており、研究者が研究対象のモチーフ部位の配列をアップロードすると、その部位と相互作用する可能性が高いキナーゼのリストを受け取ることができます。
CSTの研究部門のエグゼクティブダイレクターであるSean Beausoleilは、「このツールを用いることにより、リン酸化されたタンパク質を調べ、どのキナーゼがそのリン酸化イベントに関連しているかを予測することができます。」と述べています。
このツールは、がんの発達や他の障害などの疾患を促進する、シグナル伝達経路の機能不全の調査に用いることができます。治療によって変化したリン酸化イベントに関するデータをアップロードすると、自動的にリン酸化の増加または減少に関連するキナーゼを特定することができます。
Seanは、「まさしく、リン酸化プロテオミクスの復興です。」と述べています。「リン酸化実験を行うすべての人が、このツールでデータセットの確認を行うべきでしょう。」
その他のリソース
上記の25億種類のペプチドライブラリーを用いてCantley教授と共同開発した、CSTの一連の製品であるPTMを同定するプロテオミクス抗体とキットを含むPTMScanについてはこちらをご覧ください。
参考文献
- Johnson JL, Yaron TM, Huntsman EM, et al. An atlas of substrate specificities for the human serine/threonine kinome. Nature. 2023;613(7945):759-766. doi:10.1038/s41586-022-05575-3. CC BY 4.0.
- Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science. 2002;298(5600):1912-1934. doi:10.1126/science.1075762
- Massachusetts Institute of Technology. Enzyme 'atlas' helps researchers decipher cellular pathways: Biologists have mapped out more than 300 protein kinases and their targets, which they hope could yield new leads for cancer drugs. ScienceDaily. ScienceDaily, 12 January 2023
- Nita-Lazar A, Saito-Benz H, White FM. Quantitative phosphoproteomics by mass spectrometry: past, present, and future. Proteomics. 2008;8(21):4433-4443. doi:10.1002/pmic.200800231
- Han X, Aslanian A, Yates JR 3rd. Mass spectrometry for proteomics. Curr Opin Chem Biol. 2008;12(5):483-490. doi:10.1016/j.cbpa.2008.07.024